Epilepsy and Seizure
PsychoGenics offers a variety of translational EEG approaches including electrographic (EEG) and behavioral identification of seizures. We offer custom electrode configurations for EEG, ECoG, and local field potential (LFP) recordings from cortical and subcortical regions using our scalable tethered or telemetric (wireless) platforms for which we record up to 48 rats or mice simultaneously.
Many progressive neurologic diseases such as epilepsy require preclinical animal models that slowly develop the disease in order to test interventions at various stages of the disease process. PGI uses synchronized Video – EEG to record spontaneous or chemically-induced seizures. We have studied generalized and focal seizures in transgenic models of rare disease epilepsies including (A) Tsc1flox/flox-GFAP-Cre (Tsc1GFAPCKO) mice (model of Tuberous Sclerosis), (B) the kainic acid-induced mesial temporal lobe epilepsy (MTLE) model of refractory epilepsy, (C) B6.129P2(C)-Mecp2tm1.1Bird/J Het mice (model of Rett syndrome), and (D) B6(Cg) – Scn1atm1.1Dsf/J mice (model of Dravet). Considering the strong construct and etiological validity of the models and the robustness of the seizures, drug screens using genetic models provide strong translation to the clinic.
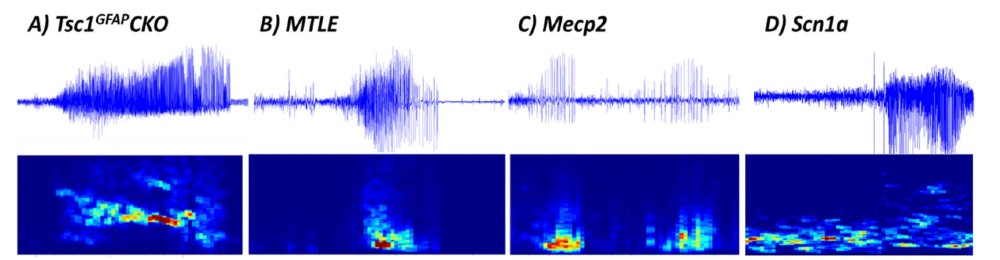
Figure 1: Exemplar electrographic seizures for the different models of epilepsy (raw EEG traces) (top). Spectrogram (raw power spectral density or PSD) (bottom) also reveals clear transient deviations from background EEG activity. All graphs have x-axis as 60sec and y-axis as -150-250μV (TSC),-250-250μV (MTLE)-30-60μV (Mecp2),-350-350μV (Scn1a) and 0-40Hz for the relative power spectrograms. EEG allows detection of multifaceted epileptiform activity including individual spikes or sharp waves, paroxysmal spike-and-wave activity, slow waves, and spike bursts or trains.
Tuberous Sclerosis
Work funded by the Tuberous Sclerosis Alliance (TSA) on behalf of the TSC Preclinical Consortium has enabled prioritization of potential new treatments based on comparing head-to-head data using consistent animal models and testing procedures in specific TSC mouse models, including the Tsc1GFAPCKO mice, which develop stereotypical electrographic seizures identified by EEG (Figure 1A) at around 1-2 months of age and became progressively more frequent with age. Rapamycin has strong efficacy for preventing seizures and prolonging survival in Tsc1GFAPCKO mice (Figure 2).
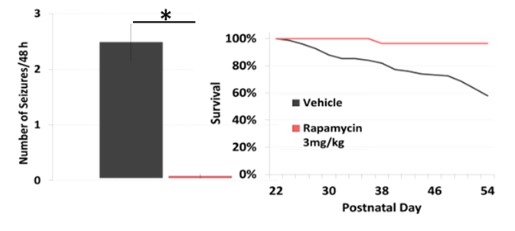
Figure 2: Effects of rapamycin on electrographic seizure count (Left Panel) and survival (Right Panel).Treatment started at P21.
Temporal Lobe Epilepsy
A model of refractory epilepsy is the intrahippocampal kainate mouse model of mesial temporal lobe epilepsy (MTLE). Reproducable hippocampal discharges (HDs) emerge within 2-3 weeks after kainic acid injection (Figure 3) and offer a
reliable model for anti-epilepsy drug testing.
Western blotting can be used to monitor protein levels, post-translational modifications, and to understand the mechanism of action for drug candidates. Protein abundance is evaluated by in lane normalization to a housekeeping protein, when possible.
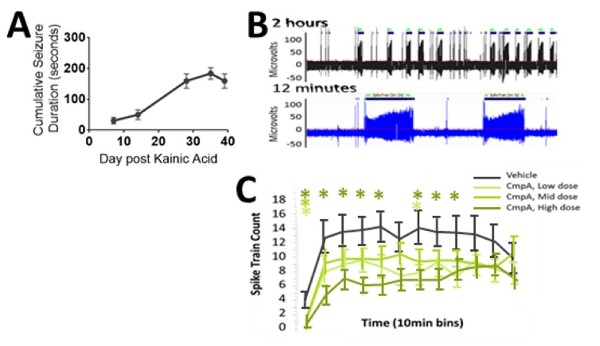
Figure 3. (A) Hippocampal Discharges emerge steadily over 2-3 weeks when they reach a plateau optimal for drug testing. (B) Characteristic large amplitude ictal and low amplitude interictal activity is seen throughout the recording allowing for quantitative measurement of drug activity related to the PK of the compound as seen in (C); note the time of dosing occurs immediately prior to the first ten minute bin).
Rare Epilepsy Syndromes
Genetic models of rare epilepsy syndromes are important in the discovery of novel treatments and for the study of the varied underlying pathologies of genetic epilepsies. Characterization of the seizures and the sensitivity to treatment is paramount in establishing validity of the models.
Mecp2tm1.1Bird/J mouse model of Rett
Rett syndrome, caused most commonly by a mutation in MECP2, presents with epilepsy and often exhibits multiple seizure types. Mecp2 tm1.1Bird/J Het female mice, regarded as a useful model for its high construct validity, may also offer face validity due to its development of spontaneous cortical polyspike discharges.
B6(Cg)-Scn1atm1.1Dsf/J mouse model of Dravet
Dravet syndrome is associated with a mutation in the SCN1A gene and presents with multiple seizure types. Heterozygous conditional knock-in mice (B6(Cg)-Scn1atm1.1Dsf/J) carrying a missense mutation in SCN1A gene express the Dravet Syndrome/SMEI-associated mutation A1783V in the presence of Crerecombinase, and exhibit Dravet-like phenotypes including spontaneous seizures.
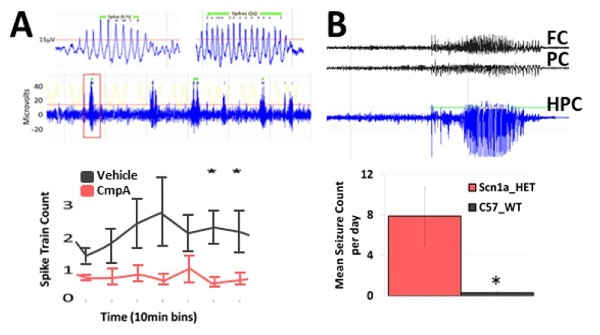
Figure 4: Spontaneous seizure activity in (A) B6.129P2(C)-Mecp2tm1.1Bird/J Het female mouse and (B) B6(Cg)-Scn1atm1.1Dsf/J Het male mouse. These mouse models develop spontaneous recurrent seizures with clear spike trains and are sensitive to potential AEDs.